超初心者向け!!RNA-seq解析シリーズ④HISAT2でマッピングする
本記事は近々以下のURLに引っ越します。
ブックマークお願いします!!
誰でもできるRNA-seq解析シリーズ!
今回はRNA-seq解析のメインとも言えるマッピングを行なっていきます!
↓これまでの記事はこちら↓
lifesciencehack-ai.hatenablog.com
HISAT2のインストール
まずはHISAT2をインストールします。
すでにhomebrewをインストールしましたので、
簡単です。
brew install プログラム名でインストールできます。
installの前後は半角スペースです。
brew tap brewsci/bio brew install hisat2
問題なくインストールされたら、以下のコマンドを入力してみましょう。
hisat2 -h
HISAT2のヘルプが表示されたらOKです。
もし、hisat2 command not foundが出たら、
インストールがうまくできていません。
リファレンスゲノムの取得
HISAT2のダウンロードが完了したら、次はリファレンスに用いるゲノム配列を用意します。
今回はマウスのRNA-seqデータでしたので、マウスのゲノム配列が必要になります。
各自解析するRNA-seqデータの動物種を予め確認しましょう。
HISAT2のサイトからリファレンスゲノムをダウンロード
さて、今回はマウスのリファレンスゲノムを取得していきます。
HISAT2でマッピングするには、リファレンスゲノムを取得した後、
マッピングの速度を上げるために、リファレンスゲノムをindex化する必要があります。
よく使われるリファレンスについては、HISAT2のサイトにindex化されたものが既にありますので、
そこからindex済のリファレンスをダウンロードしていきます。
リンク → https://ccb.jhu.edu/software/hisat2/index.shtml
画像にありますように、HISAT2のサイトの右側中段ぐらいにあります。
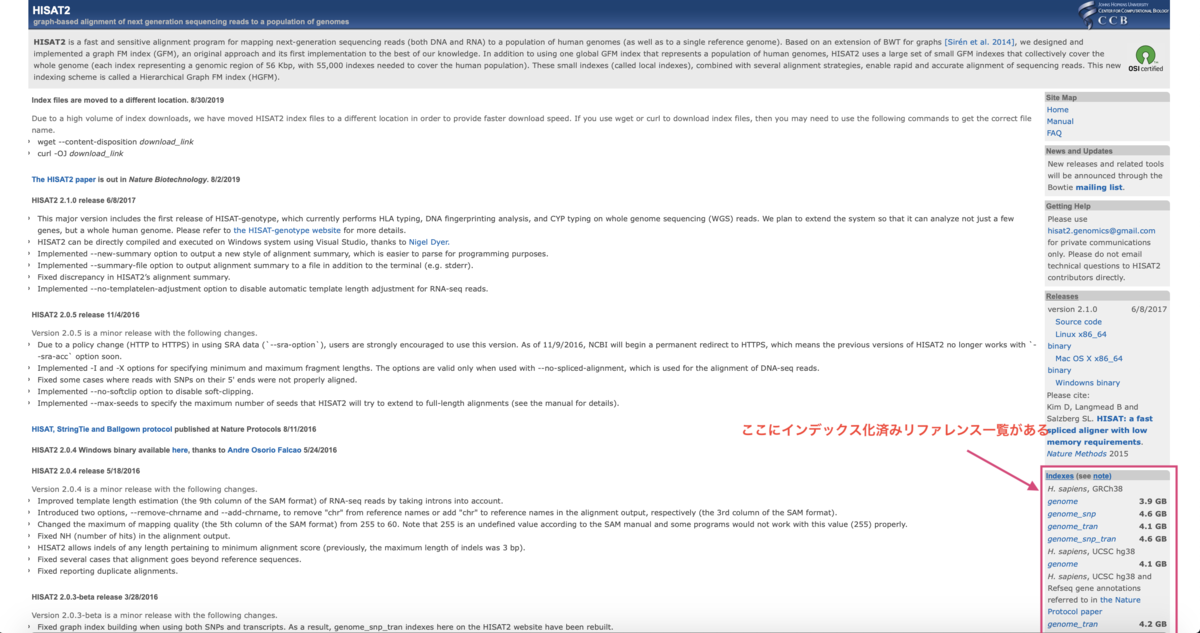
その中から、今回はマウスのリファレンスゲノムとしてよく使われるmm10をダウンロードします。
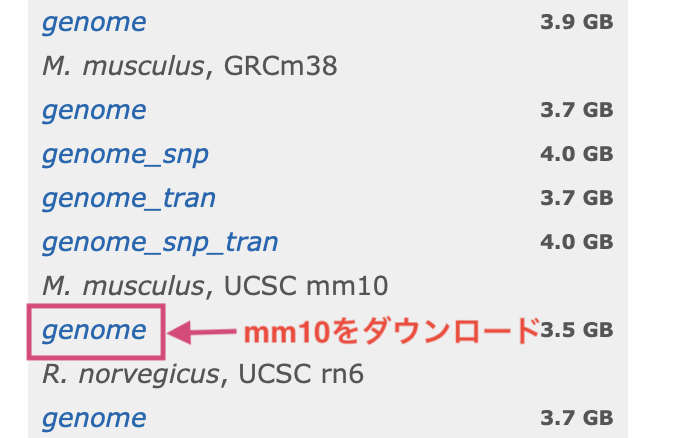
上記画像のリンクをクリックでもダウンロードできますし、
HISAT2サイトからのmm10のダウンロードは以下のコマンドをターミナルに打ち込み実行できます。
実行の前に、ディレクトリはちゃんと移動しましょう!!
ダウンロード後に、tar -zxvfコマンドでtar.gzを解凍します。
#ftpサーバーからmm10のダウンロード wget ftp://ftp.ccb.jhu.edu/pub/infphilo/hisat2/data/mm10.tar.gz #tar.gzファイルの解凍 tar -zxvf mm10.tar.gz
マッピング
では、マッピングしていきます。
その前に、先ほど作成されたリファレンスゲノムのフォルダの位置を移動させます。
画像のように、リファレンスゲノムのフォルダと解析するRNA-seqデータを同じフォルダに入れます。
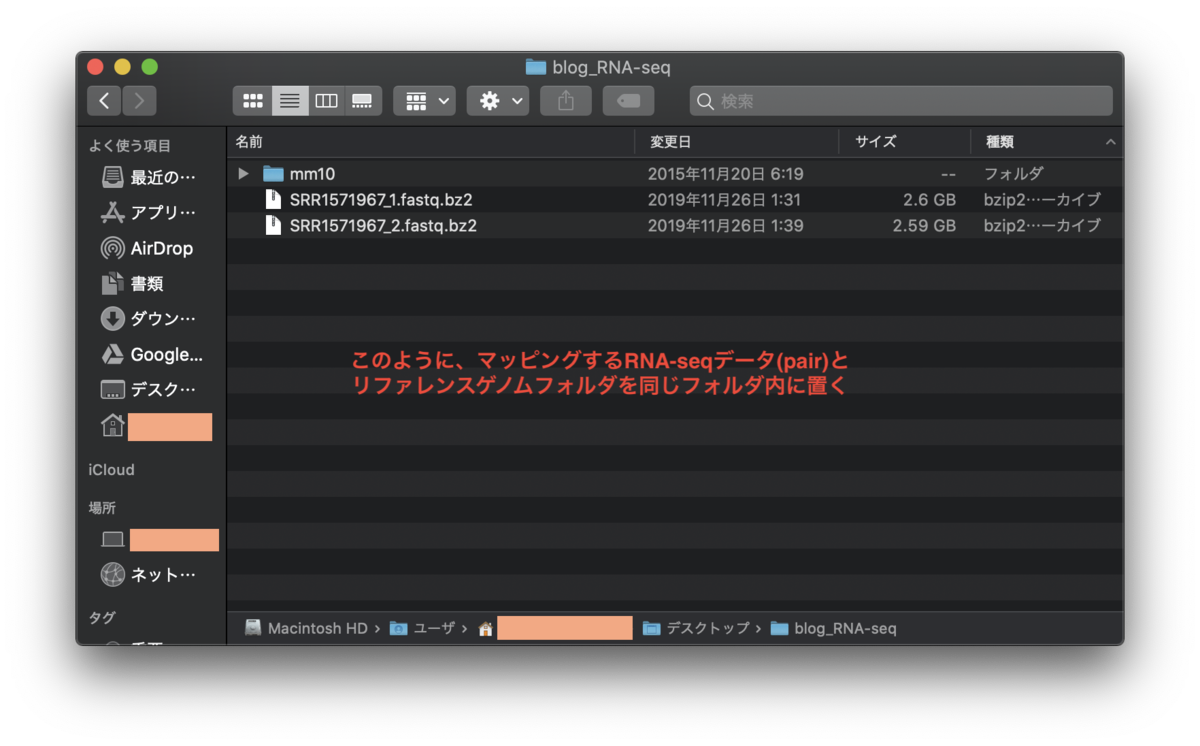
そして、ターミナルの作業ディレクトリをRNA-seqとリファレンスの入ったディレクトリに移動させましょう。
#ディレクトリの移動 cd /RNA-seqデータとリファレンスゲノムフォルダのあるディレクトリ ls mm10 SRR1571967_1.fastq.bz2 SRR1571967_2.fastq.bz2
マッピングの実行
以下のコマンドを実行し、マッピングを行います。
hisat2 -t -p 4 -x mm10/genome -1 SRR1571967_1.fastq.bz2 -2 SRR1571967_2.fastq.bz2 -S SRR1571967.sam
macOSのバージョンによっては、エラーがでるかもしれません。
その際の対処法はまた別の記事にまとめたいと思います。
少々お待ち下さい。
コマンドの説明
hisat2 hisat2の実行を命令
-t 時間経過を表示
-p 4 使用するCPUのスレッド数
-x mm10/genome リファレンスゲノムファイルを指定
-1 SRR1571967_1.fastq.bz2 pair-endの1つ目のファイルを指定
-2 SRR1571967_2.fastq.bz2 pair-endの2つ目のファイルを指定
-S SRR1571967.sam samファイルで出力することと出力ファイル名を指定
結果の確認
#マッピング終了後 Time loading forward index: 00:00:07 Time loading reference: 00:00:01 Multiseed full-index search: 00:44:05 34523088 reads; of these: 34523088 (100.00%) were paired; of these: 3020396 (8.75%) aligned concordantly 0 times 28397973 (82.26%) aligned concordantly exactly 1 time 3104719 (8.99%) aligned concordantly >1 times ---- 3020396 pairs aligned concordantly 0 times; of these: 147825 (4.89%) aligned discordantly 1 time ---- 2872571 pairs aligned 0 times concordantly or discordantly; of these: 5745142 mates make up the pairs; of these: 3508209 (61.06%) aligned 0 times 1930162 (33.60%) aligned exactly 1 time 306771 (5.34%) aligned >1 times 94.92% overall alignment rate Time searching: 00:44:07 Overall time: 00:44:14
SRR1571967のマッピングの結果です。
これを全てのサンプルに対して同様に実行します。
次回は、StringTieの使い方を説明いたします。
● 超初心者向け!!RNA-seq解析シリーズ⑤StringTieの使い方