Google ColaboratoryだけでRNA-seq解析を終わらせるシリーズです。
前回までにcolaboratoryの設定とファイルの準備を行いました。
lifesciencehack-ai.hatenablog.com
今回は、RNA-seqのキモともなるマッピングをgoogle colaboratoryで実行していきます。
前回の記事でも書きましたが、google driveが100GBないと厳しいです。
- 流れ
- 0. フォルダの準備
- 1. fastqのダウンロード
- 2. HISAT2でmapping
- 3. samtoolsでソーティングとバイナリファイルへの変換
- 4. stringtieでannotation付与
- 5. Mergeファイルの作成
- 6. Ballgown用ファイルの作成
流れ
- fastqデータのダウンロード
- HISAT2でmapping
- samtoolsでsamファイルの並び替えとバイナリ化(.bamへ変換)
- stringtieでannotation付与 (.gtf)
- stringtieで各サンプルのgtfファイルをmergeする
- stringtieでballgown用ファイルの作成
1.-4. までは各サンプルに対して行い、
5. は全サンプルを4. まで終了した後、全サンプルからできたgtfファイルを使って、mergeします。
その後、そのmergeファイルを使って、各サンプルのballgown用ファイルを作成します。
0. フォルダの準備
まずはGoogle driveをマウントする。
前回の記事でのファイル準備とGoogle driveのマウントが完了すると、下の写真のような状態になっていると思います。
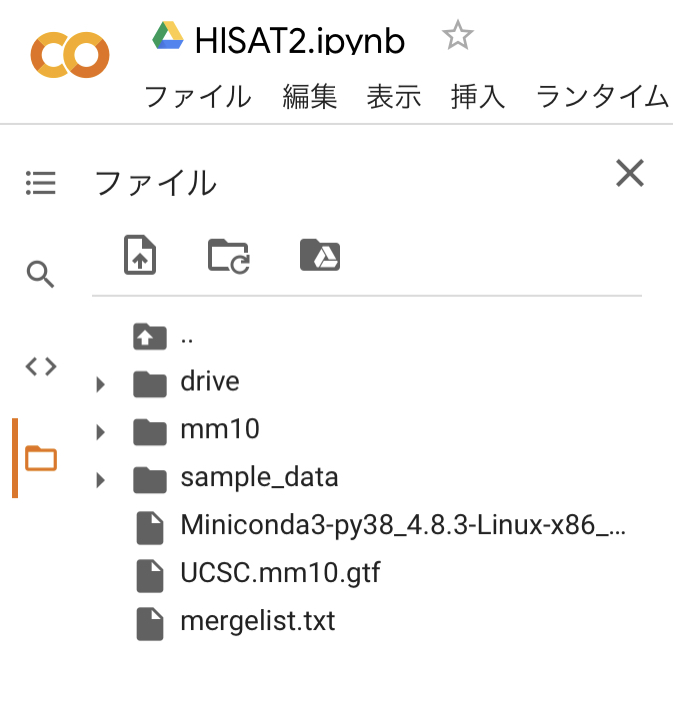
長丁場となりますので、途中でColaboratoryがシャットダウンした時のために、
マッピング後にできたファイルはGoogle driveに避難させておいた方が良いです。
Drive上に以下のコマンドで保存用フォルダを作成しましょう。
%%bash mkdir /content/drive/MyDrive/colab_RNA-seq #mergelist.txtとUCSC.mm10.gtfをdriveに移動 cp mergelist.txt /content/drive/MyDrive/colab_RNA-seq cp USCS.mm10.gtf /content/drive/MyDrive/colab_RNA-seq
前回作ったmergelist.txtとUCSC.mm10.gtfファイルですが、colab_RNA-seqフォルダを作業ディレクトリとして使用するため、colab_RNA-seqフォルダに移動しました。
Google drive開いて1番上に、
colab_RNA-seqというフォルダができているはずです。
また、ここにmergelist.txtとUCSC.mm10.gtfも格納しました。
今後作成していく、bamファイルやgtfファイルはここに入れていきます。
これで準備は終了です。
1. fastqのダウンロード
今回解析するのは、本ブログの「超初心者向け!!RNA-seq解析シリーズ」で使用したものと同じサンプルを用います。
まずはfastqファイルをダウンロードします。
%%bash #fileのダウンロード wget -c ftp://ftp.ddbj.nig.ac.jp/ddbj_database/dra/fastq/SRA183/SRA183322/SRX698161/SRR1571967_1.fastq.bz2 wget -c ftp://ftp.ddbj.nig.ac.jp/ddbj_database/dra/fastq/SRA183/SRA183322/SRX698161/SRR1571967_2.fastq.bz2
pair-endですので、1サンプルにつき2つのfasqファイルがあります。
Google colaboratory上のディレクトリ/content上にダウンロードされているはずです。
2. HISAT2でmapping
%%bash #HISAT2によるマッピング hisat2 -t -p 2 -x mm10/genome -1 SRR1571967_1.fastq.bz2 -2 SRR1571967_2.fastq.bz2 -S SRR1571967.sam --summary-file SRR1571967.txt #ファイルの削除 rm SRR1571967_1.fastq.bz2 SRR1571967_2.fastq.bz2
ちなみに、1時間半くらいかかりました。
保存容量に限りがあるので、不要なfastqファイルはどんどん削除していきましょう。
3. samtoolsでソーティングとバイナリファイルへの変換
%%bash #samtoolsによるバイナリへの変換とソーティング samtools sort -@ 2 -O bam -o SRR1571967.sort.bam SRR1571967.sam #sam fileの削除 rm SRR1571967.sam
4. stringtieでannotation付与
%%bash stringtie SRR1571967.sort.bam -o SRR1571967.gtf -p 2 -G UCSC.mm10.gtf -l SRR1571967 #Google Driveに保存する #gtfファイルの保存 cp SRR1571967.gtf /content/drive/MyDrive/RNAseq #bamファイルの保存 cp SRR1571967.sort.bam /content/drive/MyDrive/RNAseq #colabratory上からファイルを削除 rm SRR1571967.gtf SRR1571967.sort.bam
bamファイルは後ほどまた使いますので、ちゃんとdrive上に保存します。 ここまでの過程を全サンプルに対して実施します。
5. Mergeファイルの作成
1.-4. までの工程をすべてのサンプルに対して行います。
各サンプルのgtfファイルとbamファイルが1つづつGoogle driveに保存されていたらOKです。
mergeファイルを作成します。
この操作はサンプル毎に行う必要はなく、まとめて1回で終了です。
%%bash cd /content/drive/MyDrive/colab_RNA-seq stringtie —merge -G UCSC.mm10.gtf -o stringtie_merged.gtf mergelist.txt
6. Ballgown用ファイルの作成
保存フォルダの作成
最初のみです。
%%bash cd /content/drive/MyDrive/colab_RNA-seq mkdir ballgown
ballgownファイルの作成
%%bash cd /content/drive/MyDrive/colab_RNA-seq stringtie SRR1571967.sort.bam -e -B -p 2 -G stringtie_merged.gtf -o ballgown/SRR1571967/SRR1571967_ballgown.gtf
これも全てのbamファイルに対して実行します。
全部が完了すると下の画像のようなファイルが完成しているはずです。
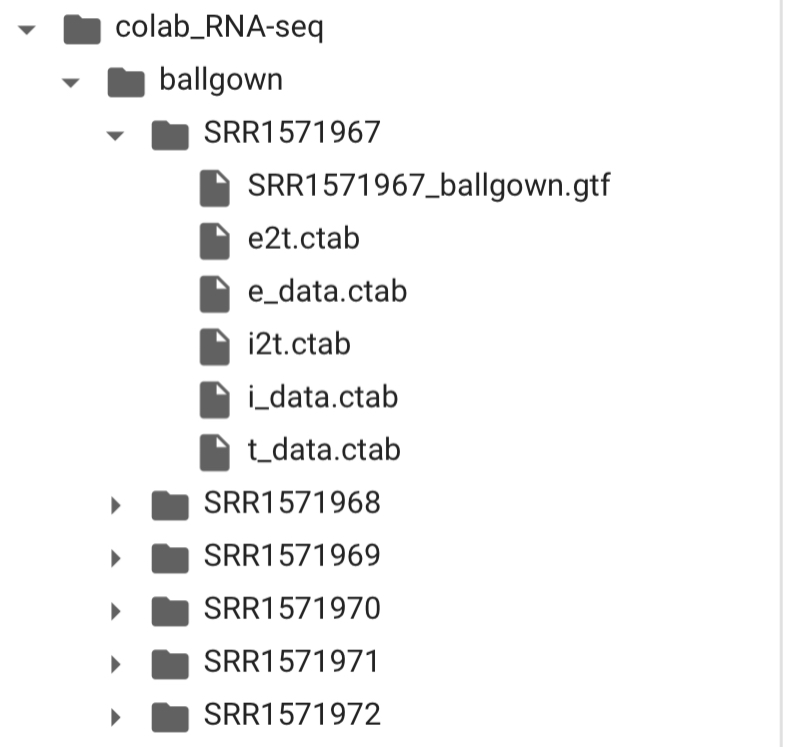
代表例としてSRR1571967のフォルダを展開していますが、
すべてのフォルダにおいて同様のファイルが作成されていれば完了です。
次回は需要はないと思いますが、Colaboratory上でRをつかってballgownによる解析を行いたいと思います。